TAMU-3DRC: AI-Guided Academic Drug Discovery
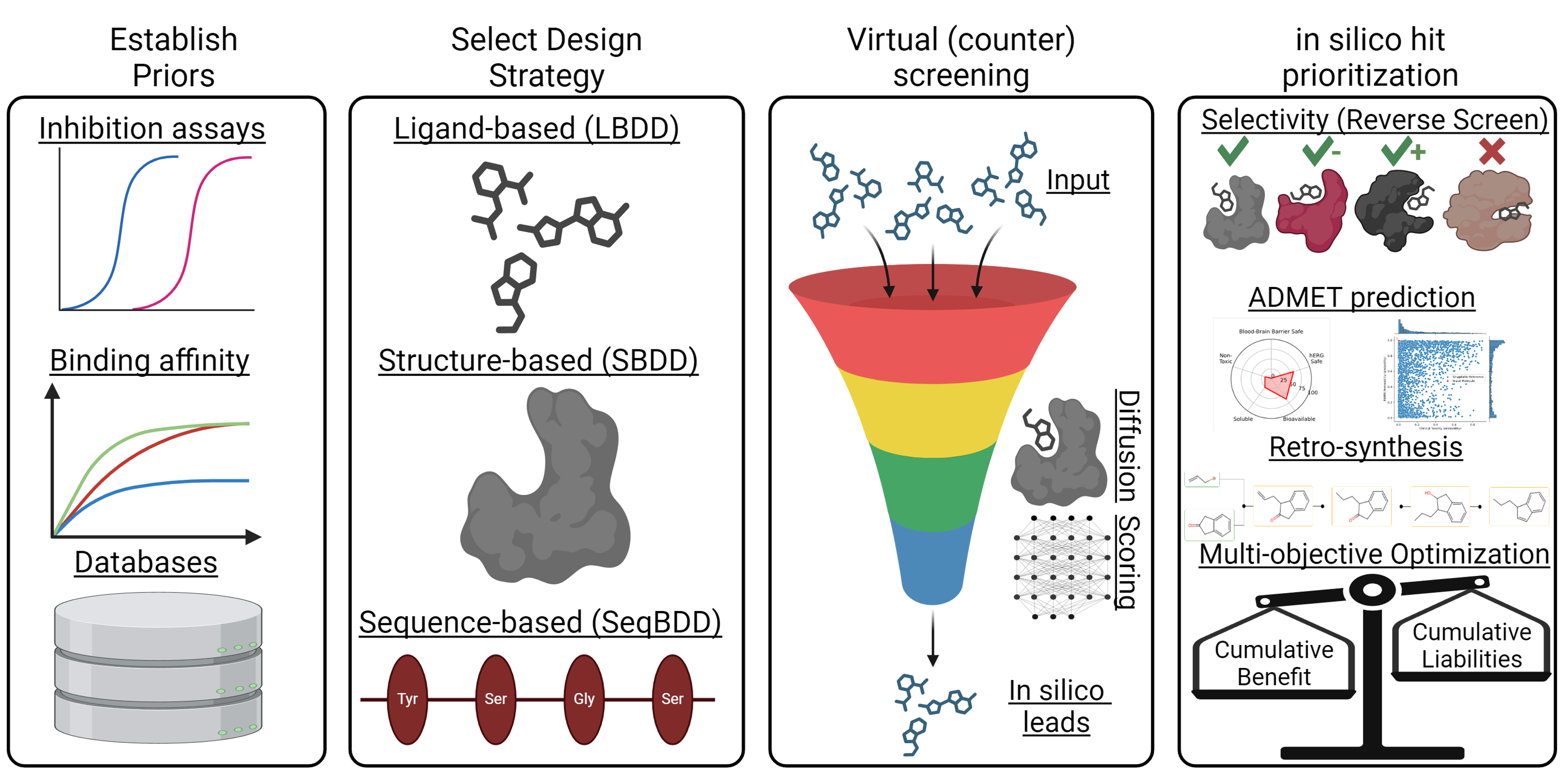
At TAMU-3DRC, we specialize in modular, computation-first discovery strategies that bring together artificial intelligence and deep domain expertise. Our interdisciplinary team includes chemists, structural biologists, machine learning scientists, and informaticians who design flexible workflows for probe and preclinical lead development.
We support both hypothesis-driven and exploratory projects through a service model that emphasizes academic-friendly integration and open-source science.
Strategic Project Planning
Our process starts with a consultation and target evaluation—assessing tractability, data readiness, and downstream feasibility. We are developing an intelligent, multi-agent system to assist in triaging projects, matching needs to the right technology stack and instrumentation, and aligning resources across the GCC-iDDD network.
Guided AI, Not Just Automation
All AI and machine learning workflows are informed by expert scientists in the loop. From feature selection to score weighting, domain knowledge shapes how we build and deploy predictive models.
We use BIOVIA Pipeline Pilot to automate complex workflows—integrating structure prediction, descriptor generation, virtual screening, and ML-SAR analysis into repeatable and auditable pipelines.
Our In Silico Discovery Stack
🧬 Structure Prediction and Binding Models
- AlphaFold2 / ESMFold: Protein structure prediction
- RFAA: All-atom folding with Rosetta refinement
- HighFold2: High-accuracy folding for macrocyclic peptides with noncanonical residues
- DynamicBind: Ligand-induced folding and flexibility
- Boltz2: Binding affinity prediction using Boltzmann-weighted scoring ensembles
🔍 Tiered Virtual Screening
We screen libraries from billions down to thousands using a tiered approach:
- 1B+ Compounds
- PharmacoNet for pharmacophore-based prescreening
- 500M Compounds
- ADMET-AI: Predictive filtering for drug-like properties
- Expert filtering, diversity clustering
- ≤1M Compounds
- GNINA: GPU-accelerated docking and scoring
- Selectivity and counter-screening
Post-screening steps include re-expansion, Boltz2-based rescoring, and SAR optimization by our medicinal chemistry team.
🧪 Generative Chemistry & Library Design
- POLYGON: VAE-based generative chemistry, retrained on Enamine and MCE libraries
- SyntheMol: RL + MCTS with Chemprop models trained as Boltz2 surrogates
- SPMM: Bidirectional pretraining for molecule/property generation
These tools support de novo design, polypharmacology, and first-in-class chemistry.
🛠️ Hit-to-Lead Support
- SciFinder: Commercial synthesis planning and IP search
- AiZynthFinder: AI-powered retrosynthesis
- Custom Chemprop workflows: Reaction condition prediction, synthetic route scoring
🧬 Macrocyclic & Peptide Modalities
We are expanding efforts in macrocyclics, peptidomimetics, and mini-protein design:
- HighFold2 and Boltz2 for conformational and ensemble modeling
- BindCraft: Binding-guided sequence optimization for peptide ligands
Why Work With Us?
- 🧠 Diverse Expertise: Domain-guided AI powered by structural biology, cheminformatics, and drug design
- 🛠️ Modular Infrastructure: Automated, scalable, and open science–oriented pipelines
- 🤝 Academic Accessibility: Designed to support hypothesis-driven and exploratory research
- 🧬 End-to-End Solutions: From virtual design to synthesis planning and hit evaluation
We’re Looking for Collaborators
We welcome partnerships in:
- Cancer Therapeutics
- Neurodegenerative Disease
- Inflammation & Immunology
- Phenotypic and Target-Based Screens
- Dual Modality and Multivalent Ligand Discovery
If you’re ready to explore your target or phenotype computationally, let’s talk.